

MicroGenomics Symposium 2014 — Aims and Topics
14-16 May 2014
«Les Cordeliers» Campus, 15 rue de l’Ecole de Médicine, Paris 75006, France
Today, one of the biggest challenges for biologists is the ability to understand and analyze the genome expressed in a given cell type in the tissue environment, which means you can specifically isolate these cells and work on very small quantities of biological material. The aim of the congress is to bring together internationally renowned experts in the field and offer an overview of the present knowledge for obtaining high quality molecules and future developments in “omic” tools (DNA, RNA and protein) to genome analysis and its expression at the cell level. The latest advances in technology in the field will be introduced (flow cytometry, laser capture microdissection, DEParray, NGS, digital PCR, WGA, LC-MS/MS, RPLA, etc).
| The symposium is divided into five sessions introduced by keynote lecturers and experts in their field: | |
| 1 – Sampling methods (flow cytometry, laser capture microdissection, DEP array) 2 – Microgenomics and DNA 3 – Microgenomics and RNA 4 – Microgenomics and microRNA & NGS 5 – Microgenomics and Proteins |
|
| The Scientific Committee: | |
|
Claudia Bevilacque (INRA France) Marc Dalod (CNRS France) Charles Decraene (Institut Curie, France) Christoph Klein (University of Regensburg, Germany) Mikael Kubista (TATAA Biocenter, Czech Republic) Bénédicte Langellier (INRA France) |
Lance Liotta (George Mason University, USA) Ken Livak (Fluidigm Corporation, USA) Patrice Martin (INRA France) Michael W. Pfaffl (Technical University of Munich, Germany) Cathérine Taragnat (INRA France) |
| A selection of the recorded talks at MicroGenomics 2014: Session 1 – Sampling methods (flow cytometry, laser capture microdissection, DEP array) |
|
 |
DeCIPHEr critical steps in a flow cytometry experiment to ensure reproducible and high quality microgenomic datatsets Hervé Luche, Immuno-Phenotyping Module, Inserm, France Abstract: DeCIPHEr critical steps in a flow cytometry experiment to ensure reproducible and high quality microgenomic datatsets LUCHE H1, HADJEM L1, MELLO M1, GRENOT P1, MALISSEN B1,2,3,4, MALISSEN M1,2,3,4 1. The Centre for ImmunoPHEnomics – CIPHE, INSERM/US012, AMU, CNRS/UMS3367; 2. Centre d’Immunologie de Marseille-Luminy (CIML), Aix-Marseille University, UM2, Marseille, France; 3. Institut National de la Santé et de la Recherche Médicale (INSERM), U1104, Marseille, France; 4. Centre National de la Recherche Scientifique (CNRS), UMR7280, Marseille, France. Recent years have witnessed the growth of functional genomics projects focusing on the use of the laboratory mouse as a model of human disease. The single gene approach has lasted, and we are now entering a new phase where scientists will think in terms of networks and pathways. The Centre for ImmunoPHEnomics – CIPHE (INSERM/US012, AMU, CNRS/UMS3367) is a new institute dedicated to phenogenomics studies. With its cuttingedge expertise in mouse genetics and immunology, the CIPHE aims to develop and analyze in a massively parallel and standardized mode mouse KO/KI models allowing the understanding of the function of the mouse immune system under normal and infectious conditions. The main investigation technique of the CIPHE immune-phenotyping module is cytometry. By combining our expertise in flow cytometry and knowledge in immunology, we establish high content immunophenotyping panels (>14C) to investigate all cell subsets of the hematopoietic lineage in the mouse. Thanks to the release of a broad range of bright new reagents and the availability of high-end instruments able to monitor up to 18 fluorescent parameters at the single cell level, HC-MFC could emerge and allow the entry into a cytomic era. The Immgen experience and studies we performed in collaboration with CIML demonstrate that high content multiparameter flow cytometry (HC-MFC) was instrumental in transcriptomic studies on minute populations of highly purified cells. However, setting up HC-MFC experiments is a real technical challenge and each experimental step needs to be carefully examined and controlled to ensure meaningful analysis. In light of our contributive work to the Immgen2 consortium, we will go through all the critical steps in a flow cytometry and cell sorting experiment that need to be considered to ensure reproducible and high quality microgenomic datasets. |
 |
Microgenomics and Epistemology Thomas Heams, INRA, Génétique Animale et Biologie Intégrative, Jouy-en -Josas; AgroParisTech, Département Sciences de la Vie et Santé, Paris, France Abstract: Microgenomics and Epistemology HEAMS T INRA, Génétique Animale et Biologie Intégrative, Domaine de Vilvert Bâtiment, Jouy-en -Josas; AgroParisTech; Département Sciences de la Vie et Santé, PARIS, France. By unraveling unexpected amounts of cellular diversity, microgenomics challenges some theoretical pilars of experimental biology. For decades, cellular diversity in multicellular organisms has been explained within a conceptual deterministic framework, explaining the high level of homogeneity within cell types, as well as differences between them. Here, reproducibility is achieved by precise regulation systems, and variation stems from different uses of the same genomic information, mediated by different signals. This successful view encompasses a large number of observations but also faces severe epistemic hurdles. First, it mostly explains the origins of variations by a reductio ad infinitum: variations come from previous external variations, resulting in order originating from previous order. Second, it relies on a puzzling evolutionary dichotomy opposing competitive interactions among unicellular organisms, and cooperation between cells of multicellular organisms. In addition, single-cell studies and microgenomics have shown that, even within tissues, individual cells display various unpredictable behaviors, spanning from background noise to actual biological parameters (Kaern et al, 2005). These techniques have also described an extensive range of variations in genome sequence among supposedly clonal cells. Thus, experimental facts not only enrich our view of multicellular dynamics, but also question the very bases which we usually rely on to infer biological explanations. By taking advantage of variations over average values, microgenomics has a key role in addressing some major aspects of this ongoing debate. It could quantify how much probabilistic cellular dynamics can coexist with deterministic ones, and could disentangle genomic, transcriptomic and proteomic origins of phenotypical variations. In doing so, it could document to what extent elementary disorders can contribute to biological order (Kupiec, 2009). References : Kaern M, Elston TC, Blake WJ, and Collins JJ 2005. Stochasticity in gene expression: from theories to phenotypes. Nature Review Genetics 6(6), 451-64. Kupiec JJ 2009. The Origins of Individuals. World Scientific, Singapore. |
| Session 2 – Microgenomics and DNA | |
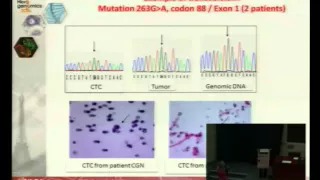 |
Single cell genetic analysis helps validating cytopathological identification of CTC in patients with Clear Cells Renal Carcinoma Patrizia Paterlini-Brechot, France Abstract: Single cell genetic analysis helps validating cytopathological identification of CTC in patients with Clear Cells Renal Carcinoma PATERLINI BRECHOT P1, BEN NJIMA B2, HOFMAN P3, HOFMAN V3, ILIE M3, CHAABOUNI H4, DOGHRI R5, BEN ROMDHANE K5 1University Paris Descartes and Inserm, Paris, France, 2Laboratory of Human Genetics, Medical Faculty of Tunis,Tunisia, 3CHU Nice and Inserm, France, 4University of Tunis, Tunisia, 5National Institute of Cancerlology, Tunis,Tunisia. Introduction – Cytopathological identification of circulating tumor cells (CTC) is a critical issue in non-invasive predictive oncology. However, in very rare cases, such as kidney cancers, the diagnostic value of cytology is weak. We thus planned to study the impact of genetic molecular analysis in combination with cytology in the CTC field. To this aim, we have taken advantage from the model of Clear Cell Renal Carcinoma (CCRC), which is characterized by the VHL genetic marker, and used genetic analysis in addition to the cytopathological study of CTC. Materials and Methods – We treated by ISET the blood of 30 patients with CCRC, collected before surgery, to isolate rare circulating cells. Cytopathological analysis was performed blindly by 3 pathologists on the isolated cells to identify CTC and distinguish them from circulating cells with uncertain malignant features (CC-UMF) and from cells with benign features (CC-BF). CTC, CC-UMF and CC-BF were then individually microdissected. Single cell DNA was preamplified, amplified by VHL-specific primers and analyzed by sequencing. VHL-specific genetic analysis was performed blindly in microdissected cells and in the corresponding tumorous tissue. Results and Discussion – We found CTC/ CC-UMF in 29/30 analyzed patients with CCRC. VHL mutations were found in the tumor of 25 out of the corresponding 29 CCRC tumors. Among 327 microdissected CTC/ CC-UMF, we obtained VHL-specific results in 205 including 64 CTC and 141 CC-UMF, according to the cytopathological analysis. VHL mutations were blindly detected in 57/64 CTC and in 125/141 CC-UMF. These results were analyzed according to the presence or absence of VHL mutations in the tumor tissue. Conclusion – This is the first study using single cell genetic analysis and cytopathology to study CTC in patients with CRCC. Our results show that genetic analysis is a very interesting approach potentially complementing cytopathology for the diagnosis of CTC. |
| Session 3 – Microgenomics and RNA | |
 |
Taking Expression Profiling to New Dimensions Mikael Kubista, TATAA Biocenter, Institute of Biotechnology AS CR in Prague, Czech Republic Abstract: High throughput single cell expression profiling KUBISTA M1,2 NOVOSADOVA V, SIDOVA M, SINDELKA R, FOROOTAN A,3 SJÖGREEN B, STÅHLBERG A,4 1 TATAA Biocenter (www.tataa.com), Gothenburg, Sweden; 2 Institute of Biotechnology, Czech Academy of Sciences, Czech Republic; 3 MultiD Analyses (www.multid.se), Gothenburg, Sweden; 4 Sahlgrenska Cancer Center, Gothenburg University, Gothenburg, Sweden. Introduction – Biological samples are complex, being composed of large number of cells of different types. When studying traditional samples containing many cells only the collective response of all the cells present is measured. However, the cells may respond differently and a small subpopulation may be critical. Today, these systems can be studied using single cell expression profiling. Here we applied single cell profiling to study the response of astrocytes to brain trauma using a mouse model. We also studied asymmetric cell division during early development of Xenopus laevis by single cell and intracellular profiling using qPCR tomography. Material and Methods – Single astrocytes from mice were enriched and collected by FACS using GFP under the control of the GFAP promoter as marker. Cells were lysed (Cellulyser, TATAA Biocenter), reverse transcribed (GrandScript, TATAA Biocenter), pre-amplified (GrandMaster PreAmp, TATAA Biocenter), and profiled using high throughput microfluidic qPCR (BioMark, Fluidigm). Data were pre-processed and cells were classified using multivariate methods (PCA, SOM, clustering) and correlation analysis with the GenEx software (ver. 6, MultiD Analysis). Results and Discussion – Astrocytes were collected from mouse brains at different time points after the induction of focal ischemia. Each cell was profiled for the expression of 47 genes. Classification revealed astrocyte reactivation with the formation of distinct subtypes (Figure). Single cell and subcellular blastomere profiling revealed asymmetric cell division is induced by asymmetric distribution of key cell fate determinants already in the fertilized cell. Conclusions – Single cell profiling is most powerful to study complex biological samples, revealing heterogeneity and to identify key expression pathways active in critical cell types. Power and robust flows for experimental and analytical analysis are available, as well as highly optimized reagents. References: M. Bengtsson, A. Ståhlberg, P. Rorsman, and M. Kubista, 2005. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Research, 15:1388. Anders Ståhlberg, Mikael Kubista, 2014. The workflow of single cell profiling using qPCR. Expert Rev. Mol. Diagn. 14(3): 321-333. |
 |
Droplet microfluidics for gene expression study at the single cell level Davide Ferraro, France Abstract: Droplet microfluidics for gene expression study at single cell level FERRARO D1, CHAMP J, TESTE B, MALAQUIN L, DESCROIX S, VIOVY JL Institut Curie UMR 168, Research Center, CNRS, UMR168, 11 rue Pierre et Marie Curie, Paris, France. Introduction – Investigating the mutated status of genes in tumor cells and how altered expression of genetic variants contributes to their development are key issues in the understanding of cancer. Standard laboratory methods are still not adapted to the isolation and quantitation of low mRNA amounts and new techniques need to be developed in particular for rare subset analysis. By reducing the volume involved, time process and the contamination risks, droplet microfluidics provide numerous advantages to perform analysis down to the single cell level1. We present here an innovative approach for the mRNA extraction and transcription at the single cell level, based on combination of droplet microfluidic and magnetic tweezers technology2. This technology has been validated in terms of sensitivity and robustness with the b-actin and HER2 transcripts. Material and Methods – mRNA purification kit based on oligo-dT beads coated was integrated in 200nL droplets to capture mRNA from total RNA. The whole analytical workflow was integrated in droplets (mRNA extraction, transcription and PCR). Final qPCR was done off-chip (Smart Cycler, Cepheid). Results and Discussion – Using this technology we were able to detect both b-Actin and HER2 genes at the single cell level. The technique was observed to be extremely stable and reproducible. Conclusions – Compare with the standard procedure, this technique allows reducing the volume by 100 times, the contamination risks and taking advantage of the high throughput droplet production. In a near future, cell lysis will be integrated in the droplet approach to integrate a complete single cell analysis while other genes of interest in cancer will be investigated. References: 1) Mary P, Dauphinot L, Bois N, Potier MC, Studer V, Tabeling P, 2011. Analysis of gene expression at the single-cell level using microdroplet-based microfluidic technology. Biomicrofluidics, 5, 024109. 2) Ali-Cherif A, Begolo S, Descroix S, Viovy JL, Malaquin L, 2012. Programmable Magnetic Tweezers and Droplet Microfluidic Device for High-Throughput Nanoliter Multi-Step Assays. Angewandte Chemie International Edition, 51:1–6. |
 |
Driving Genomics to the Single-Cell Level: Analysis of RNA Expression Using qPCR and RNAseq Ken Livak, Fellow Fluidigm Corporation South San Francisco, USA Abstract: Driving Genomics to the Single-Cell Level: Analysis of RNA Expression Using qPCR and RNA Seq LIVAK, KJ R & D, Fluidigm Corporation, South San Francisco, CA, USA Introduction – The stochastic nature of generating eukaryotic transcripts challenges conventional methods for obtaining and analyzing RNA expression data. The basic problem is how to deal with noisy data. The use of microfluidics makes it cost effective to look at a sufficient number of genes and a sufficient number of single cells to help address this noise issue. Materials and Methods – Single cells were collected either by FACS into 96-well plates or using the Fluidigm® C1™ Single-Cell Auto Prep System. qPCR was performed using 96.96 Dynamic Array™ IFCs in the Fluidigm BioMark™ HD System. For mRNA seq, libraries were sequenced using 100-bp paired end reads on an Illumina® HiSeq 2500, generating approximately four million reads for each C1 single-cell library. Results and Discussion – In an eQTL study, qPCR quantification of 92 transcripts affected by Wnt signaling in 1,440 single cells from 15 individuals identified 47 significant associations between SNP and gene expression phenotypes. This compares to finding only six SNP associations if mean expression, rather than single-cell expression, is used. This illustrates the power of using single-cell analysis to enhance the resolution of genotype-phenotype correlations. In a study of the differentiation of primary human myoblasts, a new unsupervised algorithm called Monocle was developed to analyze single-cell mRNA seq data. This algorithm can resolve the asynchrony present in a population of differentiating cells and order single cells along a developmental trajectory. Conclusions – Despite the noise inherent in single-cell transcriptomic data, analysis methods are being developed that can extract meaningful biological insights. References: Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, Sexton DW, and Holmes C, 2013. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nature Biotechnology 31:748-752. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen T, and Rinn JL, 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology doi:10.1038/nbt.2859 |
 |
Gene expression of autoreactive T cells – Biomarker and pathogenesis of Type 1 diabetes Anne Eugster, Germany Abstract: Gene expression of autoreactive T cells – Biomarker and Pathogenesis of Type 1 diabetes EUGSTER A, HENINGER A-K, KÜHN D, WILHELM C, DIETZ S, ZIEGLER A-G, BONIFACIO E Center for Regenerative Therapies Dresden and Paul Langerhans Institute Dresden, Dresden, Germany Introduction – Islet autoantibody seroconversion is the first detectable evidence of ongoing autoimmunity in the process leading to Type 1 Diabetes. While utoantibodies are markers of pre-diabetes, autoreactive T cells specific for beta-cell autoantigens are key to beta cell destruction. The BABYDIET study (1) followed children with a strong genetic predisposition for type 1 diabetes during infancy and offered the possibility to search for T cell activation against islet antigens. We used samples from this study to identify the chronical appearance of beta cell autoantigen primed CD4+ T cells and characterised their phenotype by analyzing gene expression profiles. Materials and Methods – PBMC: pre- and post seroconversion from auto-Ab negative or positive children. CFSE labeling and culturing with or without antigen for 5 days. Antigen responding CD4+ memory T cells are single cell sorted. Analysis of cytokine profiles by RT-PCR on the Fluidigm 96.96 dynamic array. Results and Discussion – We find that primed T cell responses are specific to autoantibody positive children and appear predominantly around the age of seroconversion. Comparing gene expression profiles of autoantibody positive and negative children we see that reactive cells differ already prior to seroconversion, and that they may predict the risk of autoimmunity. Evidence of regulatory responses is found in autoantibody negative – but not positive children. After seroconversion, autoantigen reactive cells develop an IFNg+ profile, differing from a vaccine responder profile. Conclusions – Gene expression profiling of antigen responsive T cells provides both biomarkers and pathogenesis discovery in Type 1 Diabetes. References: 1 Hummel S, Pflüger M, Hummel M, Bonifacio E, Ziegler AG, 2011. Primary dietary intervention study to reduce the risk of islet autoimmunity in children at increased risk for type 1 diabetes: the BABYDIET study. Diabetes Care. 34(6): 1301-5 |
| Session 4 – Microgenomics and microRNA & NGS | |
 |
Exosome isolation and holistic expression profiling using RNA-Seq and RT-qPCR Michael W. Pfaffl, Technical University of Münich, Germany Abstract: Exosome isolation and holistic expression profiling using RNA-Seq and RTqPCR PFAFFL MW AND KIRCHNER B Physiology Weihenstephan, ZIEL Research Center for Nutrition and Food Sciences,Technische Universität München, Freising, Germany Introduction – Small RNA, in particular microRNA, regulate gene expression by post transcriptional binding and thereby suppressing protein translation. They are present in most eukaryotic cells and play an important role in almost all physiological or regulative processes. Small RNA were detected in various matrices, such as blood, plasma, saliva and urine. However, very less information is available about the small RNA composition in biofluids such as milk and whether milk possesses its own defined small RNA profile differing from blood. Furthermore the small RNA transcriptome differences between whole milk versus milk exosomal isolates were investigated. Materials and Methods – To generate a holistic overview of all present small RNA Next Generation Sequencing (small RNA-Seq) was performed on whole blood, whole milk and exosomes during the first lactation days. Exosomes were purified via ultracentrifugation, due to the higher exosomal and RNA quality. Small RNA-Seq was performed using an Illumina HiSeq 2500 and subsequent data analysis was done independently, using either the GGS and GGA stations from Genomatix Software GmbH (Munich, Germany) or using freely available python scripts and R-packages (Bioconductor). First focus was on the dynamic regulation of microRNA in milk and/or exosomes in comparison to blood. Significant regulation of microRNA between different tissues and time points was defined by a fold change of expression of at least 2-fold and a Benjamini-Hochberg adjusted p-value of less than 0.05. To validate these findings key microRNA were quantified via RT-qPCR for fold change comparisons. Experimentally validated mRNA targets for regulated microRNA were taken from the Tarbase 6.0 database from Diana Lab (Athens, Greece) and pathway analyses were generated using GePS (Genomatix Pathway System). Results and Discussion – RNA Seq clearly showed that milk and exosomes possess its own unique small RNA profile compared to blood. This highlights its dynamic changes during the first lactation days. Pathway analysis for affected targets revealed significant impact on cell cycle progression, cell adhesion, DNA repair, apoptosis, and oncogenic defense. Conclusion – This study underlines the potential role of microRNA (and small RNA in general) in mammary gland physiology. Milk microRNA and exosomes seem essential for the mammary gland immune system, but also as an active modulator in the newborn calf. |
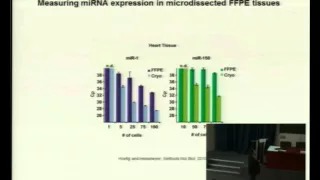 |
Measuring microRNA expression in size-limited samples Kai Peter Höfig, Munich University, Germany Abstract: Measuring MicroRNA Expression in Size-Limited Samples HOEFIG K1 AND HEISSMEYER V Research Unit Molecular Immune Regulation (AMIR), Helmholtz Zentrum München GmbH, German Research Center for Environmental Health. Marchioninistr. 25, 81377 München, Germany. Introduction – MicroRNA are small non-coding RNA of an average length of 22 nucleotides, which repress translation of a large number of target mRNA. The particular importance of this group of small RNA arises from the ever growing evidence that they control many biological processes and that deregulation of individual microRNA frequently results in cancer. The expression of microRNA is spatially and temporarily fine-tuned and expression levels can reach more than 50 000 copies of one microRNA within a single cell. It is well documented that the comparison of microRNA signatures of normal and diseased tissues results in a small number of differentially expressed microRNA, which are consequently of high diagnostic value. However, measuring microRNA expression can easily produce false-positive results, due to the high sequence similarity of the microRNA within families and because biologically inactive pre-microRNA as well as contaminating bystander cells may falsify the signal. Material and Methods – The application of a quantitative PCR-based method is described here to specifically and reliably detect microRNA expression levels from as little as 50 cells. Pure cell populations were either derived from fluorescence activated cell sorting (FACS) or from laser capture microdissection (LCM). Additionally, a combination of quantitative PCR and LCM can be applied to measure microRNA expression of cells obtained from formalin-fixed, paraffin-embedded tissues. Results and Discussion – We measured miR-155 expression of FACS-sorted germinal center T cells, derived from an immunized mouse, and compared it to mixed splenic CD4+ T cells, obtained from a non-immunized mouse. The endogenous miR-155 expression was approximately six times higher in germinal center T cells than in a mixed CD4+ T cell population. In a second approach it was demonstrated that LCM can be used to isolate cells that cannot be accessed by FACS. Conclusions – We established simple FACS and LCM-based methods to access rare cell samples of normal and diseased tissues for miRNA-profiling. References: Hoefig K and Heissmeyer V, 2010. Measuring MicroRNA Expression in Size-Limited FACS-Sorted and Microdissected Samples. Methods in Molecular Biology, 667, 47-63. |
| |
A combination of microgenomics approaches to understand the ipmpact of gut microbiota on the regulation of the hypothalamo-pituitary-adrenal axis in rats Bénédicte Langellier, France Abstract: A combination of microgenomics approaches to understand the impact of gut microbiota on the regulation of the hypothalamo-pituitary-adrenal axis in ratsi ANGLADE PA,B, LANGELIER BA,B, CRAPART NC,F, MAXIMIN EA,B, JACKSON SE, CHU SE, GERARD PA,B, MARTIN PC,D, BEVILACQUA CC,D, RABOT SA,B,1 a INRA, UMR1319 Micalis, Jouy-en-Josas, France; b AgroParisTech, Micalis, Jouy-en-Josas, France; c INRA, UMR1313 GABI, Plateforme @BRIDGe-ICE, Jouy-en-Josas, France; dAgroParisTech, GABI, Jouy-en-Josas, France; eThermo Fisher Scientific-Life Technologies, San Francisco, CA, USA; f Excilone, Elancourt, France Introduction – The gastro-intestinal tract hosts a complex microbial community, the gut microbiota, which is nowadays regarded as a full organ taking part in the host physiology. Recently, we showed in rats that the gut microbiota regulates the hypothalamo-pituitary-adrenal (HPA) axis reactivity, as reflected by a higher corticosterone systemic concentration in germfree animals than in conventional ones following an acute stress (Crumeyrolle-Arias et al, 2014). Our aim was to identify which HPA axis genes are regulated by the gut microbiota. Material and methods – We subjected germfree and conventional rats to an acute stress, killed them by decapitation and collected and froze the brain, as well as pituitary and adrenal glands. Non stressed rats served as controls. Brain and adrenal gland sections were stained to localize areas of interest, and specific cell harvesting techniques were applied, according to the distribution and density of the key cells: micro-punching of the paraventricular nucleus (PVN) in the hypothalamus, laser capture microdissection (LCM) of cell clusters in the zona fasciculata of the adrenal gland cortex. The expression level of a panel of 48 genes, selected according to their role in the HPA axis activity and reactivity, was analyzed using TaqMan® Array Micro Fluidic Cards; qRT-PCR was carried out in both tissues and a DNA pre-amplification was applied in the adrenal gland cell clusters. The development of a distinct combination of techniques is in progress for analysis of the pituitary gland: immuno-labelling to identify the corticotropic cells which are scattered in the anterior lobe of the gland, LCM of 20 to 30 single cells followed by gene expression analysis involving DNA pre-amplification with the TaqMan® PreAmp Cells-to-CT™ kit. Results and discussion – First results indicate that gut microbiota regulates the expression of several HPA axis genes in the PVN and in the zona fasciculata of the adrenal gland cortex in both non stressed and stressed rats. In conclusion, this microgenomics workflow allowed us to address the issue of gene expression analysis in a heterogeneous set of tissues involved in a specific neuroendocrine pathway. Reference: Crumeyrolle-Arias M, Jaglin M, Bruneau A, Vancassel S, Cardona A, Daugé V, Naudon L and Rabot S 2014. Psychoneuroendocrinology 42:207-217. This work was supported by Thermo Fisher Scientific-Life Technologies who kindly provided PicoPure RNA extraction kits, TaqMan® PreAmp Master Mix and Cells-to-CT™ kits and TaqMan® Array Micro Fluidic Cards. We acknowledge Excilone for supporting LCM experiments. |
 |
Comparative analysis of RNA sequencing methods for degraded or low-input samples Rahul Satija, Broad Institute of MIT, Massachusetts, USA Abstract: Comparative analysis of RNA sequencing methods for degraded or low-input samples SATIJA R 1Postdoctoral Fellow – Regev Lab, Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge MA 02142. RNA-seq is an effective method for studying the transcriptome, but it can be difficult to apply to scarce or degraded RNA from fixed clinical samples, rare cell populations or cadavers. I will present our recent comparative analysis to determine optimal protocols for low-input and low-quality RNA-seq. We created and sequenced 10 libraries using different protocols on the same human RNA sample, and developed a set of computational metrics to evaluate and benchmark different techniques. We found that the RNase H method performed best for chemically fragmented, low-quality RNA, and can even effectively replace oligo(dT)-based methods for standard RNA-seq. Moreover, the SMARTer method successfully created libraries for ultra-low inputs, enabling the development of a protocol for single cell RNA-seq. I will conclude by demonstrating how we have applied these single cell methods to characterize enormous cellular heterogeneity, reconstruct regulatory circuitry, and discover novel cellular populations in the mammalian immune system. |
 |
Low RNAseq microdissected plant tissue Bernard Dubreucq, France Abstract: Low RNAseq microdissected plant tissue BALZERGUE S1, BORREGA N , YANSOUNI J, BRUNAUD V, DELANNOY E , FAURE JD , DUBREUCQ B2 1 Unité de Recherche en Génomique Végétale (URGV), UMR INRA 1165 – Université d’Evry Val d’Essonne – ERL CNRS 8196, 2 rue Gaston Crémieux CP 5708, 91057 Evry cedex France; 2Institut Jean-Pierre Bourgin, UMR 1318 INRA-AgroParis Tech, Centre de Versailles-Grignon, Bâtiment 2 Route de St-Cyr, RD10, 78026 Versailles Cedex France, Introduction – Plants are complex organisms whose growth involves coordinated development in space and time. In order to fully understand the molecular mechanisms occurring during development, this complexity needs to taken into account Laser assisted microdissection (LAM) is a method of choice to provide access to specific cell types. In this work we have coupled LAM with RNAseq to address the questions of RNA quality for quantitative transcriptomic studies. Materials and Methods – Embedded Arabidopsis thaliana developing embryos have been microdissected using a ZEISS PALM microscope. Total RNA was extracted and quality checked. Libraries have been obtained using the SMARTer® Ultra™ Low RNA kit and RNAseq was performed using the Illumina Hiseq2000 machine. The data obtained were processed packages astqc for quality, bowtie2, samtools for reads mapping and count, and RSeQC for distribution along gene model. Results and Discussion – We used 5, 0.5 and 0.1 ng of total microdissected RNA as starting materiel. The initial quality was critical since RIN indexes below 6 did not allow successful production of libraries. The number of mapped reads was very good and almost the same in all samples. More than 16000 genes were detected using 0.1 and 5 ng of RNA template and 11200 with 0.5ng. The data show an increase of poly A/T stretches in the reads when decreasing the RNA template quantity, together with a bias in the 5’-3’ covering of the reads over gene bodies. On the same line the RNA-seq read counts reproducibility was lower for low input quantities. Conclusions – We show that it is possible to perform RNAseq on as low as 0.1 ng microdissected plant total RNA templates. We can successfully identify plant gene expression in very small cell samples using this technique, but quantification of differences does not appear to be as straightforward and will need additional studies to estimate the technical variability. |
| Session 5 – Microgenomics and Proteins | |
 |
Advantages or disadvantages of different methods for proteomics analysis Lance Liotta, George Mason University, USA Abstract: Molecular profiling using Laser Microdissection, Protein Microarrays, and Genomics, for the Individualized Therapy of Metastatic Breast Cancer LIOTTA LA, ESPINA V, PETRICOIN E, WULFKUHLE J, PIEROBON M 1University Professor, Co-Director Center for Applied Proteomics and Molecular Medicine, Medical Director Clinical Proteomics Lab, College of Science, George Mason University, 10900 University Blvd., Manassas, Virginia 20110, Metastasis is the major cause of breast cancer treatment failure. Genomic sequencing and proteomic pathway mapping of metastastic lesions has recently shown that the molecular factors driving the growth or drug resistance of metastatic colonies may be quite different from the primary tumor from which they are derived. Consequently, molecular profiling of the primary tumor may NOT accurately predict the optimal therapy that would be effective for potentially lethal metastasis. Microgenomic and microproteomic technology, including Laser Capture Microdissection (LCM), Reverse Phase Protein Micrarrays (RPPA), and Genomic Sequencing, have made it possible to conduct a new category of clinical trial to individualize the therapy of metastatic cancer. For these trials, the molecular profiling of metastatic breast lesions, not the primary tumor, is used as a starting point for individualized therapy of stage IV disease. The goal is to elucidate the deranged, mutated, or hyperactive molecular (proteomic and genomic) signaling network within metastastic lesions destined to progress in the face of currently available treatment. The hope is that gathering this information can a) provide individualized therapies that would not have been originally selected by the treating physician, and b) identify entirely new targets for therapeutic intervention. A first critically important technology used for these trials is LCM. Cellular heterogeneity of tumors hinders accurate molecular profiling for individualized therapy. For this reason, homogenization of a tumor surgical specimen or a biopsy will yield false negative and false positive data depending on the relative contribution of host cells and tumor sub-clones that are in unknown proportion in the tissue sample. LCM permits the procurement of tumor cells under direct microscopic visualization for proteomic and genomic analysis of tumor versus host (stroma) cells or phenotypically defined subpopulations of tumor cells. A second critical technology is RPPA. The phosphorylation, or activation state of kinase-driven signal networks contains important information concerning both the disease pathogenesis as well as potential for therapeutic target selection. Modulation of ongoing cellular kinase activity represents one of the most rapidly growing arenas in new drug discovery. Through the use of phospho-specific antibodies on the RPPA platform, it is now possible to quantitatively evaluate the state of entire portions of a signaling pathway or cascade, even though the cell is lysed, by looking at hundreds of signal proteins and kinase substrates at once. By measuring the proportion of those protein molecules that are phosphorylated, or activated, we can infer the level of activity of that signal node and the upstream and downstream members of the signal pathway. The activated signal pathway contains the drug target of interest. It is theoretically feasible to administer combination therapy targeting multiple interdependent points along a pathogenic pathway or targeting multiple distinct yet cooperating dysregulated pathways. The ultimate goal is tailored combination therapy of metastasis that provides higher individual efficacy with lower toxicity. Under the novel design of this trial (Side-Out Trial), a breast cancer patient’s metastatic lesion is biopsied, microdissected with LCM, and profiled with proteomic and genomic technology. A treatment selection committee reviews the patient’s data and recommends therapeutic options based on the molecular findings. Side Out Trial One is now completed. It successfully met its statistical goal of extending the progression free survival period to a ratio better than 1.3 compared to the progression free survival period for the individual patient’s last prior therapy. The therapies selected based on molecular profiling for all 25 patients were different than the physician’s choice without the profiling. This study demonstrates the feasibility of personalized cancer treatment for metastatic breast cancer using a first-of-its-kind highly multiplexed molecular profile based rationalized treatment recommendation. Based on the success of the first pilot Side Out Trial, Side out Trial number Two is now open as a multi-institutional consortium accruing patients with stage IV breast cancer. |
 |
Mass Spectrometry Imaging coupled to Microproteomics: From Imaging to identification of proteins on tissue section Maxence Wisztorski, University of Lille I, France Abstract: Mass Spectrometry Imaging coupled to Microproteomics: From identification of proteins on tissue section WISZTORSKI M, FRANCK J, QUANICO J LSMBFA, EA 4550, University Lille 1, Cité Scientifique, Bat SN3, porte 115, Villeneuve, France. Introduction – Novel and sensitive strategies such as MALDI MS imaging (MSI) allow the precise localization of endogenous compounds directly on tissue section and allow the study of interaction between cells environment [Franck 2009]. Nevertheless, strategies remains difficult and only allows access to a limited number of major proteins. Material and Methods – This presentation will briefly depict MALDI MSI through different applications such metabolites, as exogenious compounds peptides/proteins localization but also nucleic acids like mRNA. Aspect of sample preparation analysis will be exposed and strategies for in situ identification will be discuss. In particular, in order to improve proteins identification from tissue sections, we recently proposed a hybrid strategy using Liquid Micro Junction to extract tryptic peptides coming from localized on-tissue digestion followed by a LC MS & MS/MS analysis leading to the [Quanico 2013, Wisztorski 2013]. Results and Discussions – Localization of compounds coming from various source of tissue such as plants, invertebrates or mammals will exemplify the large panel of applications for which MALDI MSI can be used. We will also illustrate how recent improvements for proteins identification will open the gate of this technique for clinical applications in particular for cancer research. |
 |
Unlocking the prognostic significance of microdissected proximal lesions from human colon Daniel W. Rosenberg, University of Connecticut Health Center USA Abstract: Unlocking the prognostic significance of microdissected proximal lesions from the human colon DREW DA, DEVERS T AND ROSENBERG DW HealthNet, Inc. Chair in Cancer Biology and Professor of Medicine, Director, Colon Cancer Prevention Program, Investigator, Center for Molecular Medicine, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030-3101 USA. Despite increased implementation of screening colonoscopy, interval cancers in the proximal (right) colon remain a major public health concern. This observation underscores the limitations of current screening strategies and the critical need to develop advanced endoscopic techniques. The presence of aberrant crypt foci (ACF), the earliest morphologically identifiable epithelial abnormality in human colon, may serve as a surrogate marker for cancer risk, but the significance of this lesion within the proximal colon has not been addressed. Our laboratory has implemented high-definition chromoendoscopy on ~ 200 normal subjects to characterize the neoplastic potential of proximal ACF. The development of a combinatorial approach to maximize genomic and molecular data acquisition from ACF has been necessitated due to their exceedingly small size (< 5mm). In the following study, methods are described that combine UV/IR and laser-capture microdissection with an ultrasensitive nanofluidic proteomic immunoassay and DNA mass spectrometry. These highly sensitive methodologies enable the interrogation of the mutational spectrum of microscopic biospsies removed from the human colon and to interpret post-translational modifications of MAPK within the context of underlying oncogenic changes. Among the somatic mutations that have been identified include mutations to APCR876* and FLT3I836M, as well as an insertion/deletion with the EGFR gene, mutations that are related to the underlying pathology of the lesion. Finally, microdissection has been combined with a powerful genome-wide methylation analysis to begin to uncover the methylation state of early neoplasia within the proximal colon. |
| MicroGenomics 2014 – Online Proceedings | MicroGenomics 2014 – Poster PDFs |
|---|---|
| MicroGenomics 2014 – Event webpage | MicroGenomics 2014 – Presentation PDFs |